Access structural information, both raw diffraction data and PDB entries, for proteins that could be optimized to improve the production of advanced biofuels and bioproducts.
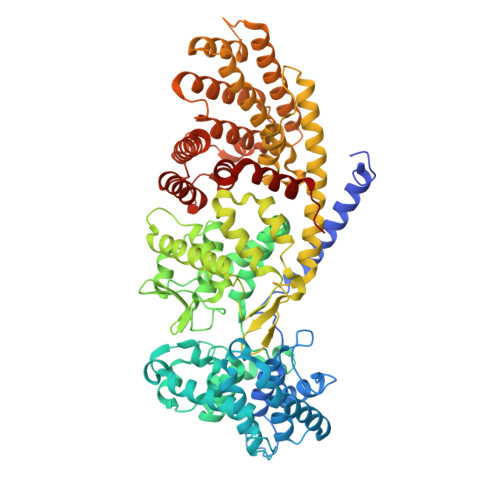
Crystal structure of Grindelia robusta 7,13-copalyl diphosphate synthase
Diterpenoid natural products serve critical functions in plant development and ecological adaptation and many diterpenoids have economic value as bioproducts. The family of class II diterpene synthases catalyzes the committed reactions in diterpenoid biosynthesis, converting a common geranylgeranyl diphosphate precursor into different bicyclic prenyl diphosphate scaffolds. Enzymatic rearrangement and modification of these precursors generates the diversity of bioactive diterpenoids. We report the crystal structure of Grindelia robusta 7,13-copalyl diphosphate synthase, GrTPS2, at 2.1 Å of resolution. GrTPS2 catalyzes the committed reaction in the biosynthesis of grindelic acid, which represents the signature metabolite in species of gumweed (Grindelia spp., Asteraceae). Grindelic acid has been explored as a potential source for drug leads and biofuel production. The GrTPS2 crystal structure adopts the conserved three-domain fold of class II diterpene synthases featuring a functional active site in the γβ-domain and a vestigial ɑ-domain. Substrate docking into the active site of the GrTPS2 apo protein structure predicted catalytic amino acids. Biochemical characterization of protein variants identified residues with impact on enzyme activity and catalytic specificity. Specifically, mutagenesis of Y457 provided mechanistic insight into the position-specific deprotonation of the intermediary carbocation to form the characteristic 7,13 double bond of 7,13-copalyl diphosphate.
Cowie AE, Pereira JH, DeGiovanni A, McAndrew RP, Palayam M, Peek JO, Muchlinski AJ, Yoshikuni Y, Shabek N, Adams PD, Zerbe P: The crystal structure of Grindelia robusta 7,13-copalyl diphosphate synthase reveals active site features controlling catalytic specificity. J. Biol. Chem. 2024PDB IDs: 9B99 [Diffraction data]
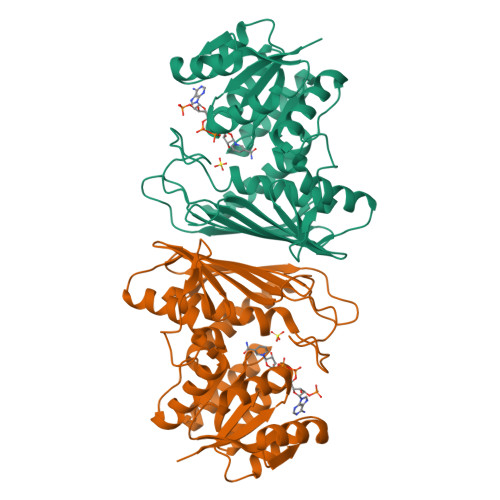
Crystal structure of CHMS Dehydrogenase PmdC from Comamonas testosteroni bound to cofactor NADP
Pyrone-2,4-dicarboxylic acid (PDC) is a valuable polymer precursor that can be derived from the microbial degradation of lignin. The key enzyme in the microbial production of PDC is 4-carboxy-2-hydroxymuconate-6-semialdehyde (CHMS) dehydrogenase, which acts on the substrate CHMS. We present the crystal structure of CHMS dehydrogenase (PmdC from Comamonas testosteroni) bound to the cofactor NADP, shedding light on its three-dimensional architecture, and revealing residues responsible for binding NADP. Using a combination of structural homology, molecular docking, and quantum chemistry calculations, we have predicted the binding site of CHMS. Key histidine residues in a conserved sequence are identified as crucial for binding the hydroxyl group of CHMS and facilitating dehydrogenation with NADP. Mutating these histidine residues results in a loss of enzyme activity, leading to a proposed model for the enzyme’s mechanism. These findings are expected to help guide efforts in protein and metabolic engineering to enhance PDC yields in biological routes to polymer feedstock synthesis.
Rodrigues AV, Moriarty NW, Kakumanu R, DeGiovanni A, Pereira JH, Gin JW, Chen Y, Baidoo EEK, Petzold CJ, Adams PD: Characterization of lignin degrading enzyme PmdC, which catalyzes a key step in the synthesis of polymer precursor 2-pyrone-4,6-dicarboxylic acid (PDC). J. Biol. Chem. 2024PDB IDs: 9AZO [Diffraction data]
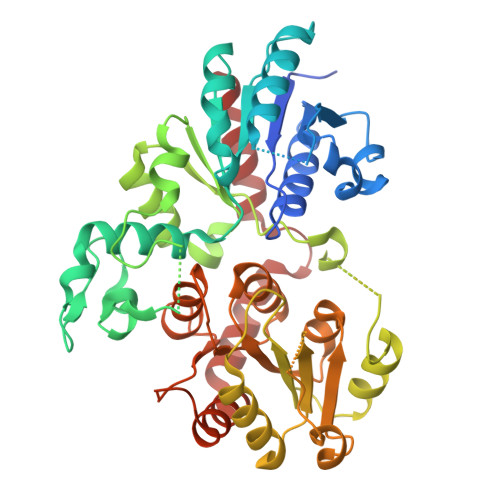
Crystal structure of the glycosyltransferase UGT95A1
Glycosylation is a predominant strategy plants use to fine-tune the properties of small molecule metabolites to affect their bioactivity, transport, and storage. It is also important in biotechnology and medicine as many glycosides are utilized in human health. Small molecule glycosylation is largely carried out by family 1 glycosyltransferases. Here, we report a structural and biochemical investigation of UGT95A1, a family 1 GT enzyme from Pilosella officinarum that exhibits a strong, unusual regiospecificity for the 3′-O position of flavonoid acceptor substrate luteolin. We obtained an apo crystal structure to help drive the analyses of a series of binding site mutants, revealing that while most residues are tolerant to mutations, key residues M145 and D464 are important for overall glycosylation activity. Interestingly, E347 is crucial for maintaining the strong preference for 3′-O glycosylation, while R462 can be mutated to increase regioselectivity. The structural determinants of regioselectivity were further confirmed in homologous enzymes. Our study also suggests that the enzyme contains large, highly dynamic, disordered regions. We showed that while most disordered regions of the protein have little to no implication in catalysis, the disordered regions conserved among investigated homologs are important to both the overall efficiency and regiospecificity of the enzyme. This report represents a comprehensive in-depth analysis of a family 1 GT enzyme with a unique substrate regiospecificity and may provide a basis for enzyme functional prediction and engineering.
Sirirungruang S, Blay V, Scott YF, Pereira JH, Hammel M, Barnum CR, Adams PD, Shih PM: Structural and biochemical basis for regiospecificity of the flavonoid glycosyltransferase UGT95A1. J. Biol. Chem. 2024, 300:107602 PDB IDs: 9BCM [Diffraction data]
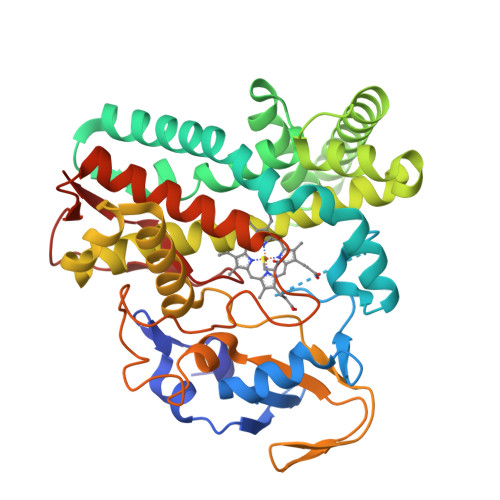
Crystal structure of an engineered P450
Biosynthesis is an environmentally benign and renewable approach that can be used to produce a broad range of natural and, in some cases, new-to-nature products. However, biology lacks many of the reactions that are available to synthetic chemists, resulting in a narrower scope of accessible products when using biosynthesis rather than synthetic chemistry. A prime example of such chemistry is carbene-transfer reactions. Although it was recently shown that carbene-transfer reactions can be performed in a cell and used for biosynthesis, carbene donors and unnatural cofactors needed to be added exogenously and transported into cells to effect the desired reactions, precluding cost-effective scale-up of the biosynthesis process with these reactions. Here we report the access to a diazo ester carbene precursor by cellular metabolism and a microbial platform for introducing unnatural carbene-transfer reactions into biosynthesis. The α-diazoester azaserine was produced by expressing a biosynthetic gene cluster in Streptomyces albus. The intracellularly produced azaserine was used as a carbene donor to cyclopropanate another intracellularly produced molecule—styrene. The reaction was catalysed by engineered P450 mutants containing a native cofactor with excellent diastereoselectivity and a moderate yield. Our study establishes a scalable, microbial platform for conducting intracellular abiological carbene-transfer reactions to functionalize a range of natural and new-to-nature products and expands the scope of organic products that can be produced by cellular metabolism.
Huang J, Quest A, Cruz-Morales P, Deng K, Pereira JH, Van Cura D, Kakumanu R, Baidoo EEK, Dan Q, Chen Y, Petzold CJ, Northen TR, Adams PD, Clark DS, Balskus EP, Hartwig JF, Mukhopadhyay A, Keasling JD: Complete integration of carbene-transfer chemistry into biosynthesis. Nature 2023, 617:403-408PDB IDs: 8FBC [Diffraction data]
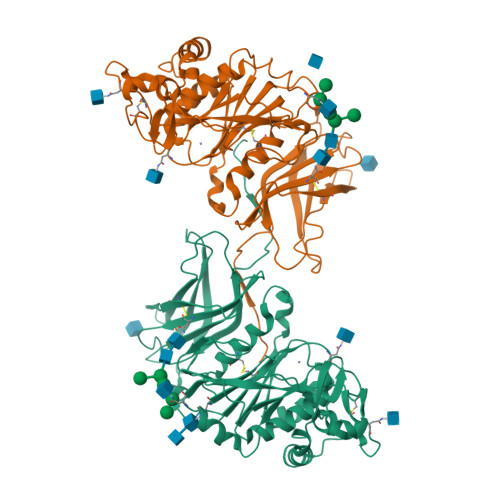
Crystal structure of GalS1 from Populus trichocarpas
Rhamnogalacturonan I (RGI) is a structurally complex pectic polysaccharide with a backbone of alternating rhamnose and galacturonic acid residues substituted with arabinan and galactan side chains. Galactan synthase 1 (GalS1) transfers galactose and arabinose to either extend or cap the β-1,4-galactan side chains of RGI, respectively. Here we report the structure of GalS1 from Populus trichocarpa, showing a modular protein consisting of an N-terminal domain that represents the founding member of a new family of carbohydrate-binding module, CBM95, and a C-terminal glycosyltransferase family 92 (GT92) catalytic domain that adopts a GT-A fold. GalS1 exists as a dimer in vitro, with stem domains interacting across the chains in a ‘handshake’ orientation that is essential for maintaining stability and activity. In addition to understanding the enzymatic mechanism of GalS1, we gained insight into the donor and acceptor substrate binding sites using deep evolutionary analysis, molecular simulations and biochemical studies. Combining all the results, a mechanism for GalS1 catalysis and a new model for pectic galactan side-chain addition are proposed.
Prabhakar PK, Pereira JH, Taujale R, Shao W, Bharadwaj VS, Chapla D, Yang JY, Bomble YJ, Moremen KW, Kannan N, Hammel M, Adams PD, Scheller HV, Urbanowicz BR: Structural and biochemical insight into a modular β-1,4-galactan synthase in plants. Nature Plants 2023, 9:486-500PDB IDs: 8D3T [Diffraction data] 8D3Z [Diffraction data]

An asymmetric disk assembly formed by tandem dimers of the tobacco mosaic viral capsid protein (TMV)
Photosynthetic light harvesting requires efficient energy transfer within dynamic networks of light-harvesting complexes embedded within phospholipid membranes. Artificial light-harvesting models are valuable tools for understanding the structural features underpinning energy absorption and transfer within chromophore arrays. Here, a method for attaching a protein-based light-harvesting model to a planar, fluid supported lipid bilayer (SLB) is developed. The protein model consists of the tobacco mosaic viral capsid proteins that are gene-doubled to create a tandem dimer (dTMV). Assemblies of dTMV break the facial symmetry of the double disk to allow for differentiation between the disk faces. A single reactive lysine residue is incorporated into the dTMV assemblies for the site-selective attachment of chromophores for light absorption. On the opposing dTMV face, a cysteine residue is incorporated for the bioconjugation of a peptide containing a polyhistidine tag for association with SLBs. The dual-modified dTMV complexes show significant association with SLBs and exhibit mobility on the bilayer. The techniques used herein offer a new method for protein-surface attachment and provide a platform for evaluating excited state energy transfer events in a dynamic, fully synthetic artificial light-harvesting system.
Dai J, Wilhelm KB, Bischoff AJ, Pereira JH, Dedeo MT, García-Almedina DM, Adams PD, Groves JT, Francis MB: A Membrane-Associated Light-Harvesting Model is Enabled by Functionalized Assemblies of Gene-Doubled TMV Proteins. Small 2023, 19:e2207805.PDB IDs: 8EAW [Diffraction data]
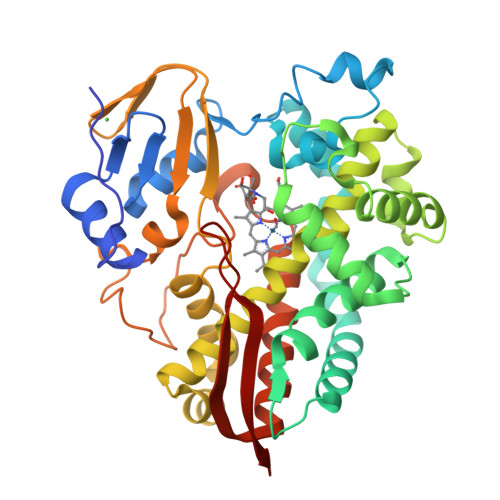
Crystal structure of cytochrome P450 enzyme CYP119 in complex with methyliridium(III) mesoporphyrin.
Artificial metalloenzymes (ArMs), which contain non-native, typically synthetic, metal cofactors, are a flourishing class of biocatalyst for unnatural reactions. Although the number of these reactions is rapidly increasing, multi-faceted mechanistic studies of ArMs comprising structural, kinetic, computational and cofactor binding data to reveal detailed mechanistic information on the effects of the protein scaffold on the structure and reactivity of ArMs are more limited. Here we report the structure of an unnatural P450 analogue using X-ray diffraction. We also report the kinetic analysis of its reaction, catalyst activation during an induction period, and the origins of the stereoselectivity for the cyclopropanation of a terpene catalysed by the iridium-containing P450 variant (Ir(Me)–CYP119). Our data reveal a mechanism initiated by the flip of the cofactor from an inactive to an active conformation. This change in conformation is followed by thousands of turnovers occurring by rate-determining formation of an iridium–carbene intermediate, thereby highlighting the influence of cofactor dynamics within a single active site on an ArM-catalysed reaction.
Bloomer BJ, Natoli SN, Garcia-Borràs M, Pereira JH, Hu DB, Adams PD, Houk KN, Clark DS, Hartwig JF: Mechanistic and structural characterization of an iridium-containing cytochrome reveals kinetically relevant cofactor dynamics. Nature Catalysis 2023, 6:39-51PDB IDs: 7UOR [Diffraction data]
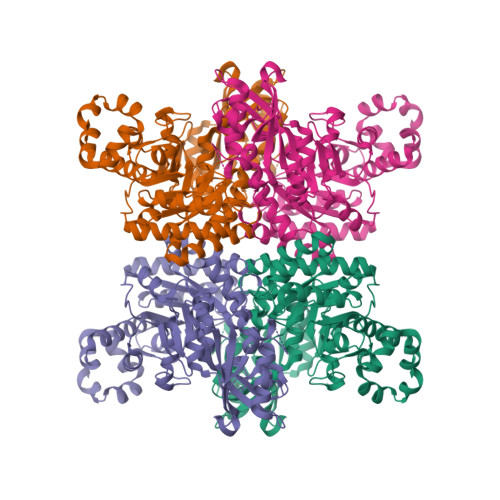
Crystal structures of RUBISCO
Oligomerization is a core structural feature that defines the form and function of many proteins. Most proteins form molecular complexes; however, there remains a dearth of diversity-driven structural studies investigating the evolutionary trajectory of these assemblies. Ribulose-1,5-bisphosphate carboxylase-oxygenase (RuBisCO) is one such enzyme that adopts multiple assemblies, although the origins and distribution of its different oligomeric states remain cryptic. Here, we retrace the evolution of ancestral and extant form II RuBisCOs, revealing a complex and diverse history of oligomerization. We structurally characterize a newly discovered tetrameric RuBisCO, elucidating how solvent-exposed surfaces can readily adopt new interactions to interconvert or give rise to new oligomeric states. We further use these principles to engineer and demonstrate how changes in oligomerization can be mediated by relatively few mutations. Our findings yield insight into how structural plasticity may give rise to new oligomeric states.
Liu AK, Pereira JH, Kehl AJ, Rosenberg DJ, Orr DJ, Chu SKS, Banda DM, Hammel M, Adams PD, Siegel JB, Shih PM: Structural plasticity enables evolution and innovation of RuBisCO assemblies. Sci. Adv. 2022, 8:eadc9440PDB IDs: 7T1C [Diffraction data] 7T1J [Diffraction data]
Structure of a newly discovered glycoside phosphorylase that makes a novel polysaccharide
The considerable utility of glycoside phosphorylases (GPs) has led to substantial efforts over the past two decades to expand the breadth of known GP activities. Driven largely by the increase of available genomic DNA sequence data, the gap between the number of sequences in the carbohydrate active enzyme database (CAZy DB) and its functionally characterized members continues to grow. This wealth of sequence data presented an exciting opportunity to explore the ever-expanding CAZy DB to discover new GPs with never-before-described functionalities. Utilizing an in silico sequence analysis of CAZy family GH94, we discovered and then functionally and structurally characterized the new GP β-1,3-N-acetylglucosaminide phosphorylase. This new GP was sourced from the genome of the cell-wall-less Mollicute bacterium, Acholeplasma laidlawii and was found to synthesize β-1,3-linked N-acetylglucosaminide linkages. The resulting poly-β-1,3-N-acetylglucosamine represents a new, previously undescribed biopolymer that completes the set of possible β-linked GlcNAc homopolysaccharides together with chitin (β-1,4) and PNAG (poly-β-1,6-N-acetylglucosamine). The new biopolymer was denoted acholetin, a combination of the genus Acholeplasma and the polysaccharide chitin, and the new GP was thus denoted acholetin phosphorylase (AchP). Use of the reverse phosphorolysis action of AchP provides an efficient method to enzymatically synthesize acholetin, which is a new biodegradable polymeric material.
Macdonald SS, Pereira JH, Liu F, Tegl G, DeGiovanni A, Wardman JF, Deutsch S, Yoshikuni Y, Adams PD, Withers SG: A Synthetic Gene Library Yields a Previously Unknown Glycoside Phosphorylase That Degrades and Assembles Poly-β-1,3-GlcNAc, Completing the Suite of β-Linked GlcNAc Polysaccharides. ACS Central Science 2022, 8:430-440PDB IDs: TBD [Diffraction data]
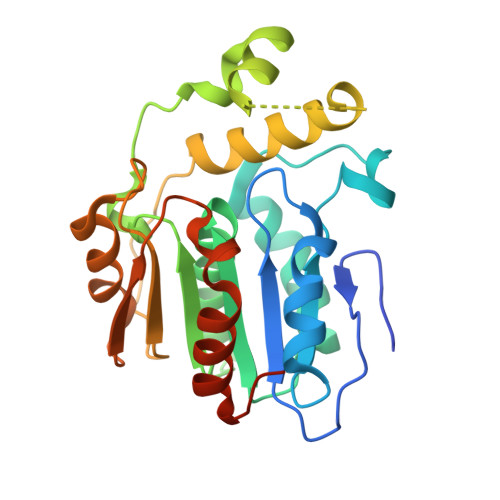
Structure of the type II thioesterase BorB from the borrelidin biosynthetic cluster
α/β hydrolases make up a large and diverse protein superfamily. In natural product biosynthesis, cis-acting thioesterase α/β hydrolases can terminate biosynthetic assembly lines and release products by hydrolyzing or cyclizing the biosynthetic intermediate. Thioesterases can also act in trans, removing aberrant intermediates and restarting stalled biosynthesis. Knockout of this “editing” function leads to reduced product titers. The borrelidin biosynthetic gene cluster from Streptomyces parvulus Tü4055 contains a hitherto uncharacterized stand-alone thioesterase, borB. In this work, we demonstrate that purified BorB cleaves acyl substrates with a preference for propionate, which supports the hypothesis that it is also an editing thioesterase. The crystal structure of BorB shows a wedgelike hydrophobic substrate binding crevice that limits substrate length. To investigate the structure–function relationship, we made chimeric BorB variants using loop regions from characterized homologues with different specificities. BorB chimeras slightly reduced activity, arguing that the modified region is a not major determinant of substrate preference. The structure–function relationships described here contribute to the process of elimination for understanding thioesterase specificity and, ultimately, engineering and applying trans-acting thioesterases in biosynthetic assembly lines.
Curran SC, Pereira JH, Baluyot MJ, Lake J, Puetz H, Rosenburg DJ, Adams P, Keasling JD: Structure and Function of BorB, the Type II Thioesterase from the Borrelidin Biosynthetic Gene Cluster. Biochemistry 2020, 59:1630-1639.PDB IDs: 6VAP [Diffraction data]
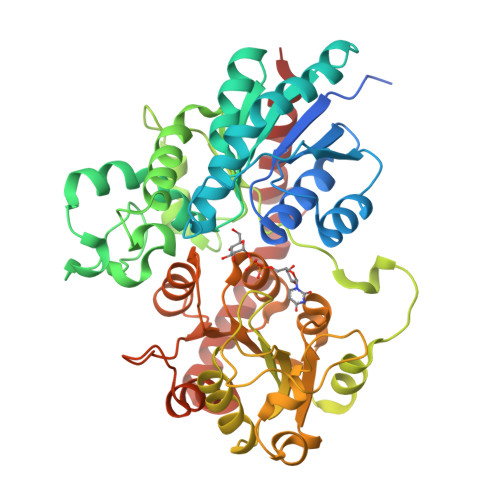
Complex between a UDP-glucosyltransferase from Polygonum tinctorium capable of glucosylating indoxyl and UDP-glucose
Glycosyltransferases (GTs) catalyze the formation of glycosidic bonds in carbohydrates and glycoconjugates, with various outcomes depending not only on the acceptor molecules they bind but also on the type of glycosidic bond they form (C–O, C–N, C–S, or C–C). Here we show that the glucosyltransferase UGT1 from the indigo plant Polygonum tinctorium catalyzes either N-, O-, or S-glycosylation with similar rates. We solve the structure of the enzyme in complex with its donor and acceptor substrates and elucidate the molecular basis of N-, O-, and S-specificities using experimental mutagenesis and QM/MM simulations, revealing distinct mechanisms for N-, O-, and S-glycosylation. We also show that the active site can be engineered to increase or favor one of the three glycosylation activities over another. These results will foster the design of more active and specific enzyme variants for production of glycosides.
Teze D, Coines J, Fredslund F, Dubey KD, Bidart GN, Adams PD, Dueber JE, Svensson B, Rovira C, Welner DH: O-/N-/S-Specificity in Glycosyltransferase Catalysis: From Mechanistic Understanding to Engineering. ACS Catalysis 2021, 11:1810-1815.PDB IDs: 6SU6 [Diffraction data]
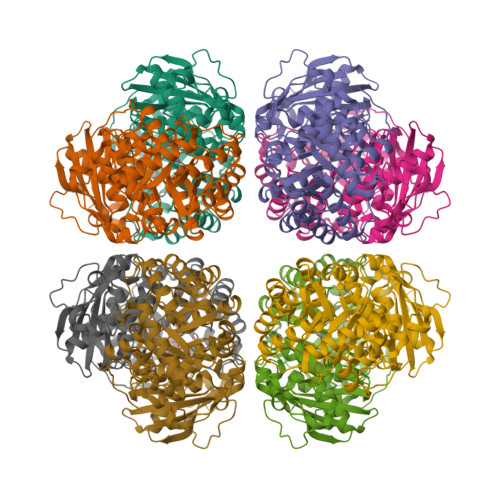
Crystal structures of RUBISCO
Rubisco sustains the biosphere through the fixation of CO2 into biomass. In plants and cyanobacteria, form I Rubisco is structurally comprised of large and small subunits, whereas all other Rubisco forms lack small subunits. The rise of the form I complex through the innovation of small subunits represents a key, yet poorly understood, transition in Rubisco’s evolution. Through metagenomic analyses, we discovered a previously uncharacterized clade sister to form I Rubisco that evolved without small subunits. This clade diverged before the evolution of cyanobacteria and the origin of the small subunit; thus, it provides a unique reference point to advance our understanding of form I Rubisco evolution. Structural and kinetic data presented here reveal how a proto-form I Rubisco assembled and functioned without the structural stability imparted from small subunits. Our findings provide insight into a key evolutionary transition of the most abundant enzyme on Earth and the predominant entry point for nearly all global organic carbon.
Banda DM, Pereira JH, Liu AK, Orr DJ, Hammel M, He C, Parry MAJ, Carmo-Silva E, Adams PD, Banfield JF, Shih PM: Novel bacterial clade reveals origin of form I Rubisco. Nature Plants 2020, 6:1158-1166PDB IDs: 6URA [Diffraction data]
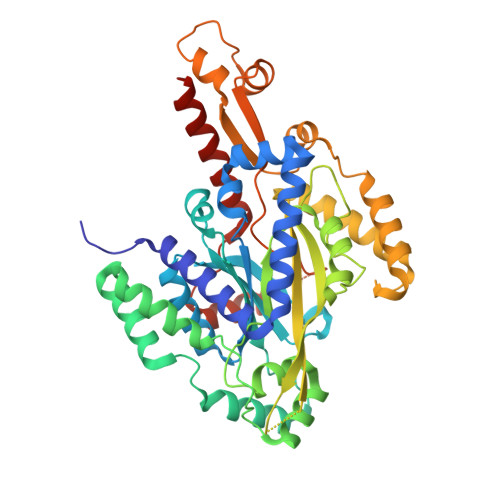
Crystal structures of hydroxyglutarate synthase
Despite intensive study, plant lysine catabolism beyond the 2-oxoadipate (2OA) intermediate remains unvalidated. Recently we described a missing step in the D-lysine catabolism of Pseudomonas putida in which 2OA is converted to D-2-hydroxyglutarate (2HG) via hydroxyglutarate synthase (HglS), a DUF1338 family protein. Here we solve the structure of HglS to 1.1 Å resolution in substrate-free form and in complex with 2OA. We propose a successive decarboxylation and intramolecular hydroxylation mechanism forming 2HG in a Fe(II)- and O2-dependent manner. Specificity is mediated by a single arginine, highly conserved across most DUF1338 proteins. An Arabidopsis thaliana HglS homolog coexpresses with known lysine catabolism enzymes, and mutants show phenotypes consistent with disrupted lysine catabolism. Structural and biochemical analysis of Oryza sativa homolog FLO7 reveals identical activity to HglS despite low sequence identity. Our results suggest DUF1338-containing enzymes catalyze the same biochemical reaction, exerting the same physiological function across bacteria and eukaryotes.
Thompson MG, Blake-Hedges JM, Pereira JH, Hangasky JA, Belcher MS, Moore WM, Barajas JF, Cruz-Morales P, Washington LJ, Haushalter RW, Eiben CB, Liu Y, Skyrud W, Benites VT, Barnum TP, Baidoo EEK, Scheller HV, Marletta MA, Shih PM, Adams PD, Keasling JD: An iron (II) dependent oxygenase performs the last missing step of plant lysine catabolism. Nat Commun. 2020, 11:2931PDB IDs: 6W1G [Diffraction data] 6W1H [Diffraction data] 6W1K [Diffraction data]

Crystal structure of acyl-ACP/acyl-CoA dehydrogenase from allylmalonyl-CoA and FK506 biosynthesis, TcsD
Terminal alkenes are easily derivatized, making them desirable functional group targets for polyketide synthase (PKS) engineering. However, they are rarely encountered in natural PKS systems. One mechanism for terminal alkene formation in PKSs is through the activity of an acyl-CoA dehydrogenase (ACAD). Herein, we use biochemical and structural analysis to understand the mechanism of terminal alkene formation catalyzed by an γ,δ-ACAD from the biosynthesis of the polyketide natural product FK506, TcsD. While TcsD is homologous to canonical α,β-ACADs, it acts regioselectively at the γ,δ-position and only on α,β-unsaturated substrates. Furthermore, this regioselectivity is controlled by a combination of bulky residues in the active site and a lateral shift in the positioning of the FAD cofactor within the enzyme. Substrate modeling suggests that TcsD utilizes a novel set of hydrogen bond donors for substrate activation and positioning, preventing dehydrogenation at the α,β position of substrates. From the structural and biochemical characterization of TcsD, key residues that contribute to regioselectivity and are unique to the protein family were determined and used to identify other putative γ,δ-ACADs that belong to diverse natural product biosynthetic gene clusters. These predictions are supported by the demonstration that a phylogenetically distant homologue of TcsD also regioselectively oxidizes α,β-unsaturated substrates. This work exemplifies a powerful approach to understand unique enzymatic reactions and will facilitate future enzyme discovery, inform enzyme engineering, and aid natural product characterization efforts.
Blake-Hedges JM, Pereira JH, Cruz-Morales P, Thompson MG, Barajas JF, Chen J, Krishna RN, Chan LJG, Nimlos D, Alonso-Martinez C, Baidoo EEK, Chen Y, Gin JW, Katz L, Petzold CJ, Adams PD, Keasling JD: Structural mechanism of regioselectivity in an unusual bacterial acyl-CoA dehydrogenase. J Am Chem Soc. 2020,142:835-846PDB IDs: 6U1V [Diffraction data]
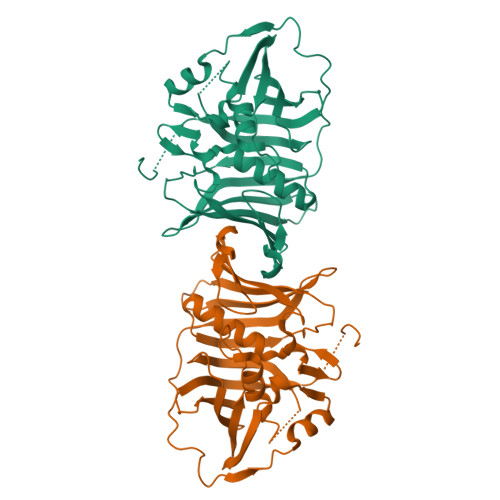
Structural insights into dehydratase substrate selection for the borrelidin and fluvirucin polyketide synthases
Engineered polyketide synthases (PKSs) are promising synthetic biology platforms for the production of chemicals with diverse applications. The dehydratase (DH) domain within modular type I PKSs generates an α,β-unsaturated bond in nascent polyketide intermediates through a dehydration reaction. Several crystal structures of DH domains have been solved, providing important structural insights into substrate selection and dehydration. Here, we present two DH domain structures from two chemically diverse PKSs. The first DH domain, isolated from the third module in the borrelidin PKS, is specific towards a trans-cyclopentane-carboxylate-containing polyketide substrate. The second DH domain, isolated from the first module in the fluvirucin B1 PKS, accepts an amide-containing polyketide intermediate. Sequence-structure analysis of these domains, in addition to previously published DH structures, display many significant similarities and key differences pertaining to substrate selection. The two major differences between BorA DH M3, FluA DH M1 and other DH domains are found in regions of unmodeled residues or residues containing high B-factors. These two regions are located between α3–β11 and β7–α2. From the catalytic Asp located in α3 to a conserved Pro in β11, the residues between them form part of the bottom of the substrate-binding cavity responsible for binding to acyl-ACP intermediates.
Barajas JF, McAndrew RP, Thompson MG, Backman TWH, Pang B, de Rond T, Pereira JH, Benites VT, Martín HG, Baidoo EEK, Hillson NJ, Adams PD, Keasling JD: Structural insights into dehydratase substrate selection for the borrelidin and fluvirucin polyketide synthases. J Ind Microbiol Biotechnol. 2019, 46:1225-1235PDB IDs: 6OBT [Diffraction data] 6OBV [Diffraction data]
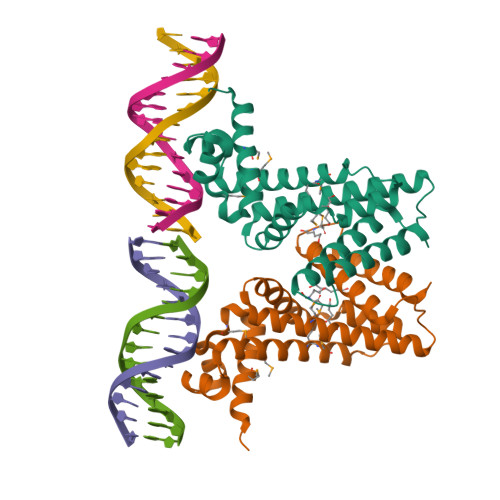
Crystal structure of EilR in complex with eilO DNA element, and effector molecules
Tightly regulated promoters are essential for numerous biological applications, where strong inducibility, portability, and scalability are desirable. Current systems are often incompatible with large-scale fermentations due to high inducer costs and strict media requirements. Here, we describe the bottom-up engineering of ‘Jungle Express’, an expression system that enables efficient gene regulation in diverse proteobacteria. This system is guided by EilR, a multidrug-binding repressor with high affinity to its optimized operator and cationic dyes that act as powerful inducers at negligible costs. In E. coli, the engineered promoters exhibit minimal basal transcription and are inducible over four orders of magnitude by 1 µM crystal violet, reaching expression levels exceeding those of the strongest current bacterial systems. Further, we provide molecular insights into specific interactions of EilR with its operator and with two inducers. The versatility of Jungle Express opens the way for tightly controlled and efficient gene expression that is not restricted to host organism, substrate, or scale.
Ruegg TL, Pereira JH, Chen JC, DeGiovanni A, Novichkov P, Mutalik VK, Tomaleri GP, Singer SW, Hillson NJ, Simmons BA, Adams PD, Thelen MP: Jungle Express is a versatile repressor system for tight transcriptional control. Nature Comm. 2018, 9:3617PDB IDs: 5VL9 [Diffraction data] 5VLG [Diffraction data] 5VLM [Diffraction data]
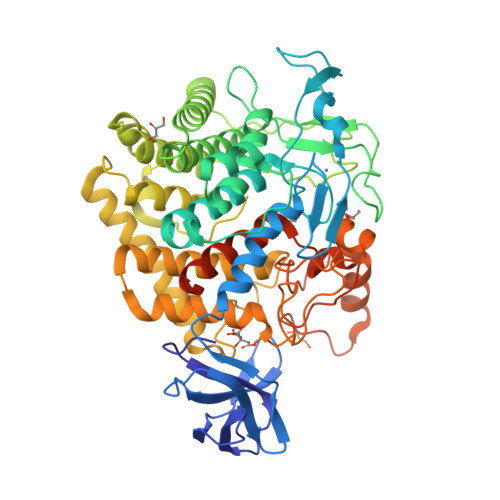
Crystal structure of wild-type and engineered GH family 9 endoglucanase J30
The development of robust enzymes, in particular cellulases, is a key step in the success of biological routes to `second-generation’ biofuels. The typical sources of the enzymes used to degrade biomass include mesophilic and thermophilic organisms. The endoglucanase J30 from glycoside hydrolase family 9 was originally identified through metagenomic analyses of compost-derived bacterial consortia. These studies, which were tailored to favor growth on targeted feedstocks, have already been shown to identify cellulases with considerable thermal tolerance. The amino-acid sequence of J30 shows comparably low identity to those of previously analyzed enzymes. As an enzyme that combines a well measurable activity with a relatively low optimal temperature (50°C) and a modest thermal tolerance, it offers the potential for structural optimization aimed at increased stability. Here, the crystal structure of wild-type J30 is presented along with that of a designed triple-mutant variant with improved characteristics for industrial applications. Through the introduction of a structural Zn2+ site, the thermal tolerance was increased by more than 10°C and was paralleled by an increase in the catalytic optimum temperature by more than 5°C.
Ellinghaus TL, Pereira JH, McAndrew RP, Welner DH, DeGiovanni AM, Guenther JM, Tran HM, Feldman T, Simmons BA, Sale KL, Adams PD: Engineering glycoside hydrolase stability by the introduction of zinc binding. Acta Cryst 2018, D74:702-710.PDB IDs: 5U0H [Diffraction data] 5U2O [Diffraction data]
Complex between a UDP-glucosyltransferase from Polygonum tinctorium capable of glucosylating indoxyl and indoxyl sulfate
Indigo is an ancient dye uniquely capable of producing the signature tones in blue denim; however, the dyeing process requires chemical steps that are environmentally damaging. We describe a sustainable dyeing strategy that not only circumvents the use of toxic reagents for indigo chemical synthesis but also removes the need for a reducing agent for dye solubilization. This strategy utilizes a glucose moiety as a biochemical protecting group to stabilize the reactive indigo precursor indoxyl to form indican, preventing spontaneous oxidation to crystalline indigo during microbial fermentation. Application of a β-glucosidase removes the protecting group from indican, resulting in indigo crystal formation in the cotton fibers. We identified the gene coding for the glucosyltransferase PtUGT1 from the indigo plant Polygonum tinctorium and solved the structure of PtUGT1. Heterologous expression of PtUGT1 in Escherichia coli supported high indican conversion, and biosynthesized indican was used to dye cotton swatches and a garment.
Hsu TM, Welner DH, Russ ZN, Cervantes B, Prathuri RL, Adams PD, Dueber JE: Employing a biochemical protecting group for a sustainable indigo dyeing strategy. Nature Chem. Biol. 2018, 14:256-261PDB IDs: 5NLM [Diffraction data]
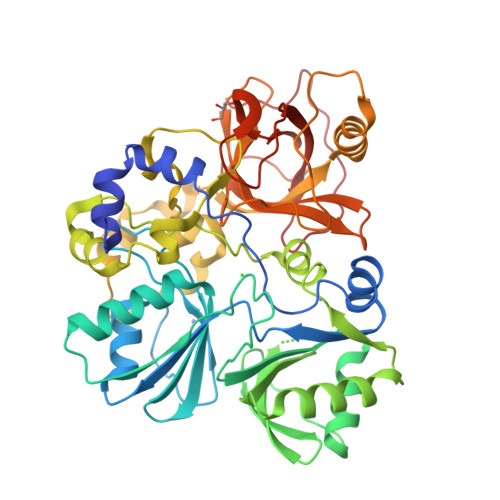
Crystal structure of Sphingomonas paucimobilis aryl O-demethylase LigM
Some strains of soil and marine bacteria have evolved intricate metabolic pathways for using environmentally derived aromatics as a carbon source. Many of these metabolic pathways go through intermediates such as vanillate, 3-O-methylgallate, and syringate. Demethylation of these compounds is essential for downstream aryl modification, ring opening, and subsequent assimilation of these compounds into the tricarboxylic acid (TCA) cycle, and, correspondingly, there are a variety of associated aryl demethylase systems that vary in complexity. Intriguingly, only a basic understanding of the least complex system, the tetrahydrofolate-dependent aryl demethylase LigM from Sphingomonas paucimobilis, a bacterial strain that metabolizes lignin-derived aromatics, was previously available. LigM-catalyzed demethylation enables further modification and ring opening of the single-ring aromatics vanillate and 3-O-methylgallate, which are common byproducts of biofuel production. Here, we characterize aryl O-demethylation by LigM and report its 1.81-Å crystal structure, revealing a unique demethylase fold and a canonical folate-binding domain. Structural homology and geometry optimization calculations enabled the identification of LigM’s tetrahydrofolate-binding site and protein–folate interactions. Computationally guided mutagenesis and kinetic analyses allowed the identification of the enzyme’s aryl-binding site location and determination of its unique, catalytic tyrosine-dependent reaction mechanism. This work defines LigM as a distinct demethylase, both structurally and functionally, and provides insight into demethylation and its reaction requirements. These results afford the mechanistic details required for efficient utilization of LigM as a tool for aryl O-demethylation and as a component of synthetic biology efforts to valorize previously underused aromatic compounds.
Kohler AC, Mills MJL, Adams PD, Simmons BA, Sale KL: Structure of aryl O-demethylase offers molecular insight into a catalytic tyrosine-dependent mechanism. Proc Natl Acad Sci U S A 2017, 114:E3205-E3214PDB IDs: 5TL4 [Diffraction data]
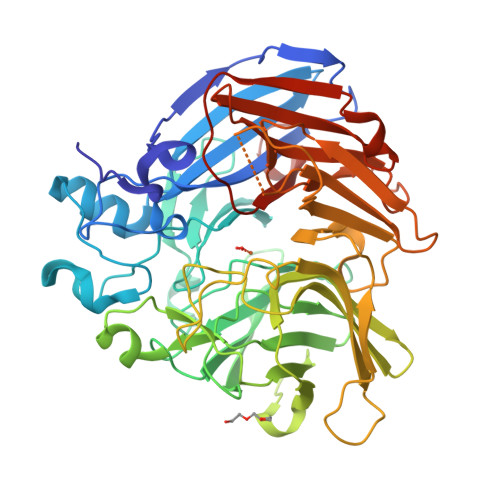
The Structure and Mechanism of NOV1, a Resveratrol-Cleaving Dioxygenase
Stilbenes are diphenyl ethene compounds produced naturally in a wide variety of plant species and some bacteria. Stilbenes are also derived from lignin during kraft pulping. Stilbene cleavage oxygenases (SCOs) cleave the central double bond of stilbenes, forming two phenolic aldehydes. Here, we report the structure of an SCO. The X-ray structure of NOV1 from Novosphingobium aromaticivorans was determined in complex with its substrate resveratrol (1.89 Å), its product vanillin (1.75 Å), and without any bound ligand (1.61 Å). The enzyme is a seven-bladed β-propeller with an iron cofactor coordinated by four histidines. In all three structures, dioxygen is observed bound to the iron in a side-on fashion. These structures, along with EPR analysis, allow us to propose a mechanism in which a ferric-superoxide reacts with substrate activated by deprotonation of a phenol group at position 4 of the substrate, which allows movement of electron density toward the central double bond and thus facilitates reaction with the ferric superoxide electrophile. Correspondingly, NOV1 cleaves a wide range of other stilbene-like compounds with a 4′-OH group, offering potential in processing some solubilized fragments of lignin into monomer aromatic compounds.
McAndrew RP, Sathitsuksanoh N, Mbughuni MM, Heins RA, Pereira JH, George A, Sale KL, Fox BG, Simmons BA, Adams PD: Structure and mechanism of NOV1, a resveratrol-cleaving dioxygenase. Proc Natl Acad Sci U S A 2016, 113:14324-14329PDB IDs: 5J53 [Diffraction data] 5J54 [Diffraction data] 5J55 [Diffraction data]
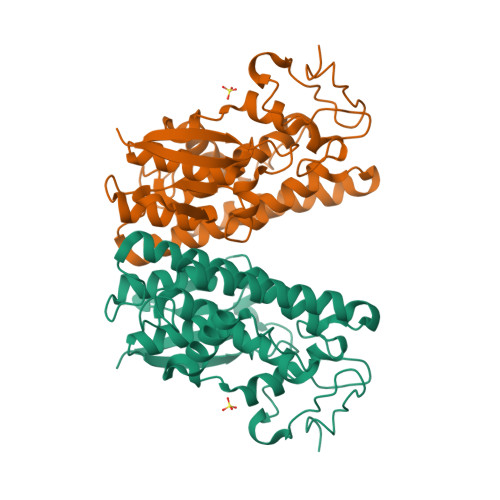
Crystal structure of LigD, LigG, LigL and LigO from Sphingobium sp. strain SYK-6
There has been great progress in the development of technology for the conversion of lignocellulosic biomass to sugars and subsequent fermentation to fuels. However, plant lignin remains an untapped source of materials for production of fuels or high value chemicals. Biological cleavage of lignin has been well characterized in fungi, in which enzymes that create free radical intermediates are used to degrade this material. In contrast, a catabolic pathway for the stereospecific cleavage of β-aryl ether units that are found in lignin has been identified in Sphingobium sp. SYK-6 bacteria. β-Aryl ether units are typically abundant in lignin, corresponding to 50–70% of all of the intermonomer linkages. Consequently, a comprehensive understanding of enzymatic β-aryl ether (β-ether) cleavage is important for future efforts to biologically process lignin and its breakdown products. The crystal structures and biochemical characterization of the NAD-dependent dehydrogenases (LigD, LigO, and LigL) and the glutathione-dependent lyase LigG provide new insights into the early and late enzymes in the β-ether degradation pathway. We present detailed information on the cofactor and substrate binding sites and on the catalytic mechanisms of these enzymes, comparing them with other known members of their respective families. Information on the Lig enzymes provides new insight into their catalysis mechanisms and can inform future strategies for using aromatic oligomers derived from plant lignin as a source of valuable aromatic compounds for biofuels and other bioproducts.
Pereira JH, Heins RA, Gall DL, McAndrew RP, Deng K, Holland KC, Donohue TJ, Noguera DR, Simmons BA, Sale KL, Ralph J, Adams PD: Structural and Biochemical Characterization of the Early and Late Enzymes in the Lignin β-aryl Ether Cleavage Pathway from Sphingobium sp SYK-6. J Biol Chem. 2016, 291:10228-38PDB IDs: 4Y98 [Diffraction data] 4Y9D [Diffraction data] 4YA6 [Diffraction data] 4YAC [Diffraction data] 4YAE [Diffraction data] 4YAG [Diffraction data] 4YAI [Diffraction data] 4YAP [Diffraction data] 4YAV [Diffraction data]
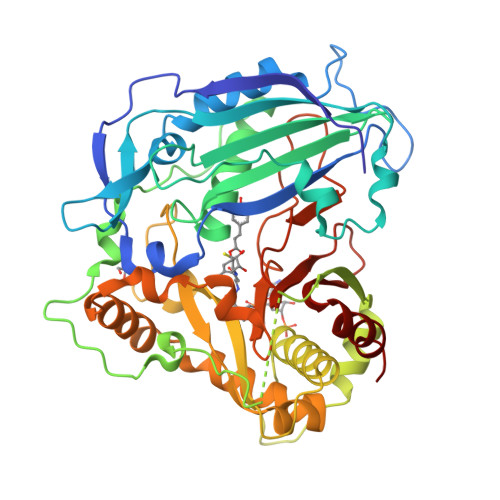
Crystal structure of PvHCT in complex with cofactors and substrates
Lignin poses a major challenge in the processing of plant biomass for agro-industrial applications. For bioengineering purposes, there is a pressing interest in identifying and characterizing the enzymes responsible for the biosynthesis of lignin. Hydroxycinnamoyl-CoA:shikimate hydroxycinnamoyl transferase (HCT; EC 2.3.1.133) is a key metabolic entry point for the synthesis of the most important lignin monomers: coniferyl and sinapyl alcohols. In this study, we investigated the substrate promiscuity of HCT from a bryophyte (Physcomitrella) and from five representatives of vascular plants (Arabidopsis, poplar, switchgrass, pine and Selaginella) using a yeast expression system. We demonstrate for these HCTs a conserved capacity to acylate with p-coumaroyl-CoA several phenolic compounds in addition to the canonical acceptor shikimate normally used during lignin biosynthesis. Using either recombinant HCT from switchgrass (PvHCT2a) or an Arabidopsis stem protein extract, we show evidence of the inhibitory effect of these phenolics on the synthesis of p-coumaroyl shikimate in vitro, which presumably occurs via a mechanism of competitive inhibition. A structural study of PvHCT2a confirmed the binding of a non-canonical acceptor in a similar manner to shikimate in the active site of the enzyme. Finally, we exploited in Arabidopsis the substrate flexibility of HCT to reduce lignin content and improve biomass saccharification by engineering transgenic lines that overproduce one of the HCT non-canonical acceptors. Our results demonstrate conservation of HCT substrate promiscuity and provide support for a new strategy for lignin reduction in the effort to improve the quality of plant biomass for forage and cellulosic biofuels.
Eudes A, Pereira JH, Yogiswara S, Wang G, Teixeira Benites V, Baidoo EE, Lee TS, Adams PD, Keasling JD, Loqué D: Exploiting The Substrate Promiscuity of Hydroxycinnamoyl-CoA:shikimate Hydroxycinnamoyl Transferase to Reduce Lignin. Plant Cell Physiol. 2016, 57:568-79PDB IDs: 5FAL [Diffraction data] 5FAN [Diffraction data]
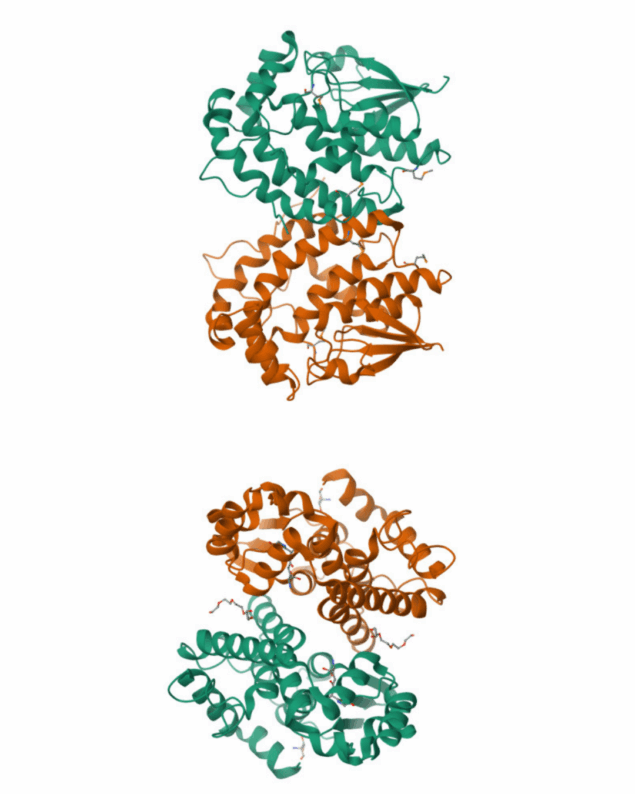
Crystal structures of LigE and LigF from Sphingobium sp. strain SYK-6
Lignin is a combinatorial polymer comprising monoaromatic units that are linked via covalent bonds. Although lignin is a potential source of valuable aromatic chemicals, its recalcitrance to chemical or biological digestion presents major obstacles to both the production of second-generation biofuels and the generation of valuable coproducts from lignin’s monoaromatic units. Degradation of lignin has been relatively well characterized in fungi, but it is less well understood in bacteria. A catabolic pathway for the enzymatic breakdown of aromatic oligomers linked via β-aryl ether bonds typically found in lignin has been reported in the bacterium Sphingobium sp. SYK-6. Here, we present x-ray crystal structures and biochemical characterization of the glutathione-dependent β-etherases, LigE and LigF, from this pathway. The crystal structures show that both enzymes belong to the canonical two-domain fold and glutathione binding site architecture of the glutathione S-transferase family. Mutagenesis of the conserved active site serine in both LigE and LigF shows that, whereas the enzymatic activity is reduced, this amino acid side chain is not absolutely essential for catalysis. The results include descriptions of cofactor binding sites, substrate binding sites, and catalytic mechanisms. Because β-aryl ether bonds account for 50–70% of all interunit linkages in lignin, understanding the mechanism of enzymatic β-aryl ether cleavage has significant potential for informing ongoing studies on the valorization of lignin.
Helmich KE, Pereira JH, Gall DL, Heins RA, McAndrew RP Jr, Bingman CA, Deng K, Holland KC, Noguera DR, Simmons BA, Sale KL, Ralph J, Donohue TJ, Adams PD, Phillips GN Jr: Structural basis of stereospecificity in the bacterial enzymatic cleavage of β-aryl ether bonds in lignin. J Biol Chem. 2015, 291:5234-46PDB IDs: 4XT0 [Diffraction data] 4YAM [Diffraction data] 4YAN [Diffraction data]
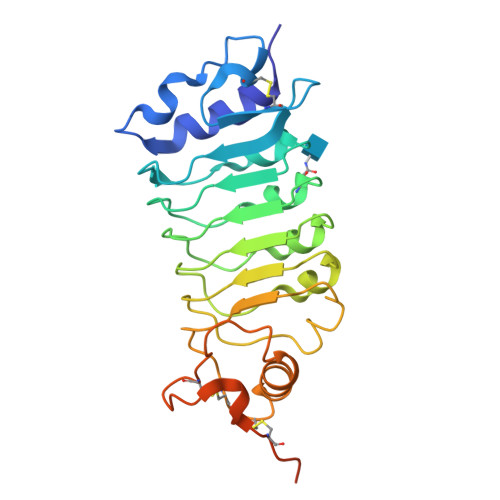
Structure of the OsSERK2 leucine rich repeat extracellular domain
Somatic embryogenesis receptor kinases (SERKs) are leucine-rich repeat (LRR)-containing integral membrane receptors that are involved in the regulation of development and immune responses in plants. It has recently been shown that rice SERK2 (OsSERK2) is essential for XA21-mediated resistance to the pathogen Xanthomonas oryzae pv. oryzae. OsSERK2 is also required for the BRI1-mediated, FLS2-mediated and EFR-mediated responses to brassinosteroids, flagellin and elongation factor Tu (EF-Tu), respectively. Here, crystal structures of the LRR domains of OsSERK2 and a D128N OsSERK2 mutant, expressed as hagfish variable lymphocyte receptor (VLR) fusions, are reported. These structures suggest that the aspartate mutation does not generate any significant conformational change in the protein, but instead leads to an altered interaction with partner receptors.
McAndrew R, Pruitt RN, Kamita SG, Pereira JH, Majumdar D, Hammock BD, Adams PD, Ronald PC: Structure of the OsSERK2 leucine-rich repeat extracellular domain. Acta Cryst. 2014, D70:3080-3086PDB IDs: 4Q3G [Diffraction data] 4Q3I [Diffraction data]
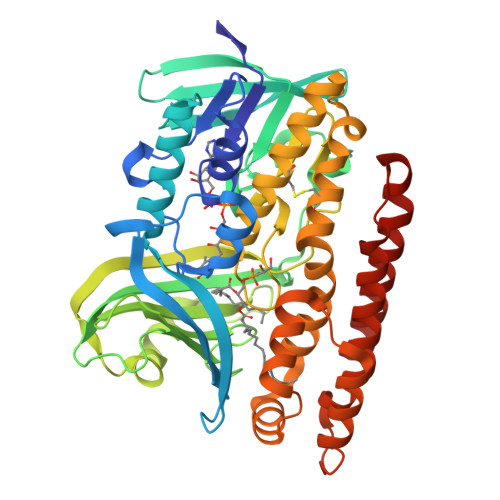
Constructing tailored isoprenoid products by structure-guided modification of geranylgeranyl reductase.
The archaeal enzyme geranylgeranyl reductase (GGR) catalyzes hydrogenation of carbon-carbon double bonds to produce the saturated alkyl chains of the organism’s unusual isoprenoid-derived cell membrane. Enzymatic reduction of isoprenoid double bonds is of considerable interest both to natural products researchers and to synthetic biologists interested in the microbial production of isoprenoid drug or biofuel molecules. Here we present crystal structures of GGR from Sulfolobus acidocaldarius, including the structure of GGR bound to geranylgeranyl pyrophosphate (GGPP). The structures are presented alongside activity data that depict the sequential reduction of GGPP to H6GGPP via the intermediates H2GGPP and H4GGPP. We then modified the enzyme to generate sequence variants that display increased rates of H6GGPP production or are able to halt the extent of reduction at H2GGPP and H4GGPP. Crystal structures of these variants not only reveal the structural bases for their altered activities; they also shed light onto the catalytic mechanism employed.
Kung Y, McAndrew RP, Xie X, Liu CC, Pereira JH, Adams PD, Keasling JD: Constructing Tailored Isoprenoid Products by Structure-Guided Modification of Geranylgeranyl Reductase. Structure 2014, 22:1028-36.PDB IDs: 4OPC [Diffraction data] 4OPD [Diffraction data] 4OPG [Diffraction data] 4OPI [Diffraction data] 4OPL [Diffraction data] 4OPT [Diffraction data] 4OPU [Diffraction data]
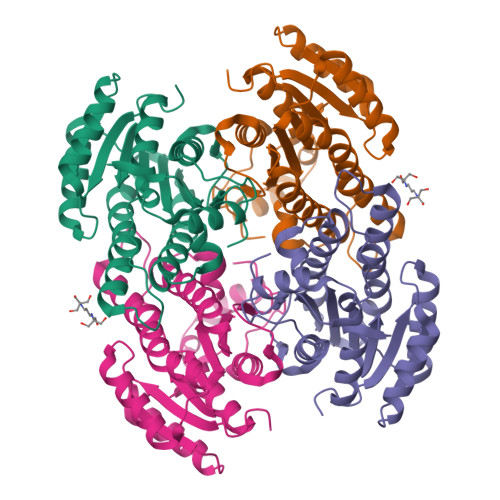
Crystal structures of FabG
Major efforts in bioenergy research have focused on producing fuels that can directly replace petroleum-derived gasoline and diesel fuel through metabolic engineering of microbial fatty acid biosynthetic pathways. Typically, growth and pathway induction are conducted under aerobic conditions, but for operational efficiency in an industrial context, anaerobic culture conditions would be preferred to obviate the need to maintain specific dissolved oxygen concentrations and to maximize the proportion of reducing equivalents directed to biofuel biosynthesis rather than ATP production. A major concern with fermentative growth conditions is elevated NADH levels, which can adversely affect cell physiology. The purpose of this study was to identify homologs of Escherichia coli FabG, an essential reductase involved in fatty acid biosynthesis, that display a higher preference for NADH than for NADPH as a cofactor. Four potential NADH-dependent FabG variants were identified through bioinformatic analyses supported by crystallographic structure determination (1.3- to 2.0-Å resolution). In vitro assays of cofactor (NADH/NADPH) preference in the four variants showed up to ∼35-fold preference for NADH, which was observed with the Cupriavidus taiwanensis FabG variant. In addition, FabG homologs were overexpressed in fatty acid- and methyl ketone-overproducing E. coli host strains under anaerobic conditions, and the C. taiwanensis variant led to a 60% higher free fatty acid titer and 75% higher methyl ketone titer relative to the titers of the control strains. With further engineering, this work could serve as a starting point for establishing a microbial host strain for production of fatty acid-derived biofuels (e.g., methyl ketones) under anaerobic conditions.
Javidpour P, Pereira JH, Goh EB, McAndrew RP, Ma SM, Friedland GD, Keasling JD, Chhabra SR, Adams PD, Beller HR: Biochemical and Structural Studies of NADH-Dependent FabG Used to Increase the Bacterial Production of Fatty Acids under Anaerobic Conditions. Appl Environ Microbiol. 2014, 80:497-505PDB IDs: 4NBT [Diffraction data] 4NBU [Diffraction data] 4NBV [Diffraction data] 4NBW [Diffraction data]
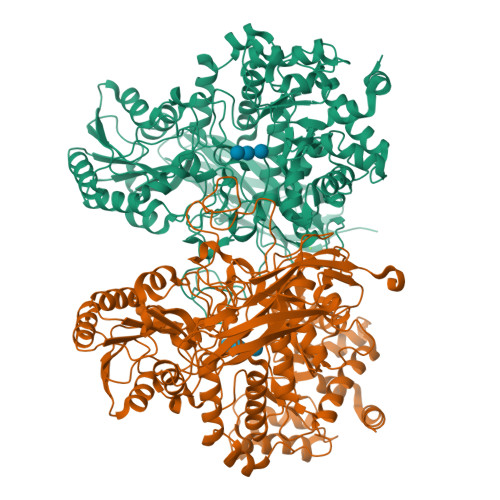
From soil to structure: a novel dimeric family 3-beta-glucosidase isolated from compost using metagenomic analysis
A recent metagenomic analysis sequenced a switchgrass-adapted compost community to identify enzymes from microorganisms that were specifically adapted to switchgrass under thermophilic conditions. These enzymes are being examined as part of the pretreatment process for the production of “second-generation” biofuels. Among the enzymes discovered was JMB19063, a novel three-domain β-glucosidase that belongs to the GH3 (glycoside hydrolase 3) family. Here, we report the structure of JMB19063 in complex with glucose and the catalytic variant D261N crystallized in the presence of cellopentaose. JMB19063 is first structure of a dimeric member of the GH3 family, and we demonstrate that dimerization is required for catalytic activity. Arg-587 and Phe-598 from the C-terminal domain of the opposing monomer are shown to interact with bound ligands in the D261N structure. Enzyme assays confirmed that these residues are absolutely essential for full catalytic activity.
McAndrew RP, Park JI, Heins RA, Reindl W, Friedland GD, D’haeseleer P, Northen T, Sale KL, Simmons BA, Adams PD: From soil to structure: a novel dimeric β-glucosidase belonging to glycoside hydrolase family 3 isolated from compost using metagenomic analysis. J. Biol. Chem. 2013, 288:14985-14992.PDB IDs: 3U4A [Diffraction data] 3U48 [Diffraction data]Raw diffraction data: 3U48, 3U4A
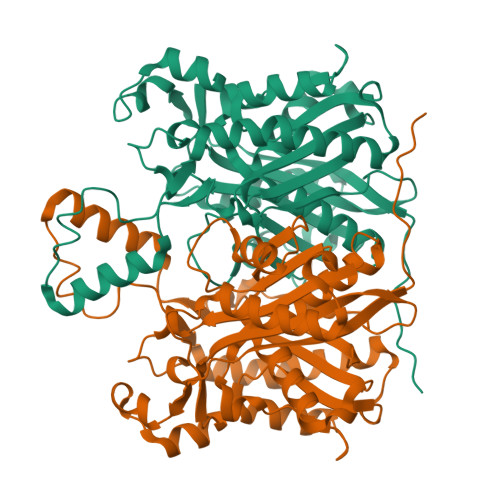
Crystal structure of FabH from Micrococcus luteus
Micrococcus luteus is a Gram-positive bacterium that produces iso- and anteiso-branched alkenes by the head-to-head condensation of fatty-acid thioesters [coenzyme A (CoA) or acyl carrier protein (ACP)]; this activity is of interest for the production of advanced biofuels. In an effort to better understand the control of the formation of branched fatty acids in M. luteus, the structure of FabH (MlFabH) was determined. FabH, or β-ketoacyl-ACP synthase III, catalyzes the initial step of fatty-acid biosynthesis: the condensation of malonyl-ACP with an acyl-CoA. Analysis of the MlFabH structure provides insights into its substrate selectivity with regard to length and branching of the acyl-CoA. The most structurally divergent region of FabH is the L9 loop region located at the dimer interface, which is involved in the formation of the acyl-binding channel and thus limits the substrate-channel size. The residue Phe336, which is positioned near the catalytic triad, appears to play a major role in branched-substrate selectivity. In addition to structural studies of MlFabH, transcriptional studies of M. luteus were also performed, focusing on the increase in the ratio of anteiso:iso-branched alkenes that was observed during the transition from early to late stationary phase. Gene-expression microarray analysis identified two genes involved in leucine and isoleucine metabolism that may explain this transition.
Pereira JH, Goh EB, Keasling JD, Beller HR, Adams PD: Structure of FabH and factors affecting the distribution of branched fatty acids in Micrococcus luteus. Acta Cryst. 2012, D68:1320-1328PDB IDs: 4EWP [Diffraction data]Raw diffraction data: 4EWP
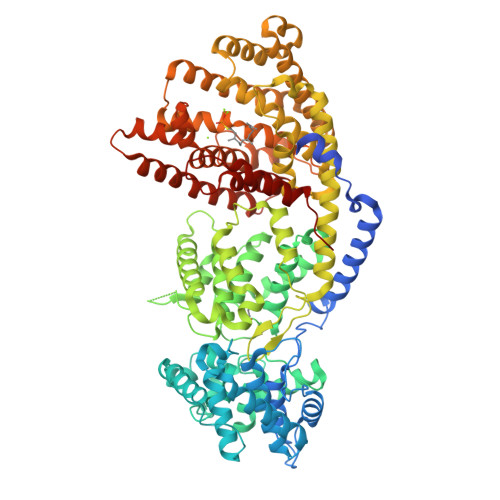
Structure of a three-domain sesquiterpene synthase: a prospective target for advanced biofuels production
The sesquiterpene bisabolene was recently identified as a biosynthetic precursor to bisabolane, an advanced biofuel with physicochemical properties similar to those of D2 diesel. High-titer microbial bisabolene production was achieved using Abies grandis α-bisabolene synthase (AgBIS). Here, we report the structure of AgBIS, a three-domain plant sesquiterpene synthase, crystallized in its apo form and bound to five different inhibitors. Structural and biochemical characterization of the AgBIS terpene synthase Class I active site leads us to propose a catalytic mechanism for the cyclization of farnesyl diphosphate into bisabolene via a bisabolyl cation intermediate. Further, we describe the nonfunctional AgBIS Class II active site whose high similarity to bifunctional diterpene synthases makes it an important link in understanding terpene synthase evolution. Practically, the AgBIS crystal structure is important in future protein engineering efforts to increase the microbial production of bisabolene.
McAndrew RP, Peralta-Yahya PP, DeGiovanni A, Pereira JH, Hadi MZ, Keasling JD, Adams PD: Structure of a Three-Domain Sesquiterpene Synthase: A Prospective Target for Advanced Biofuels Production. Structure 2011, 19:1876-1884PDB IDs: 3SAE [Diffraction data] 3SDQ [Diffraction data] 3SDR [Diffraction data] 3SDT [Diffraction data] 3SDU [Diffraction data] 3SDV [Diffraction data]Raw diffraction data: 3SDQ, 3SAE, 3SDR, 3SDT, 3SDU, 3SDV
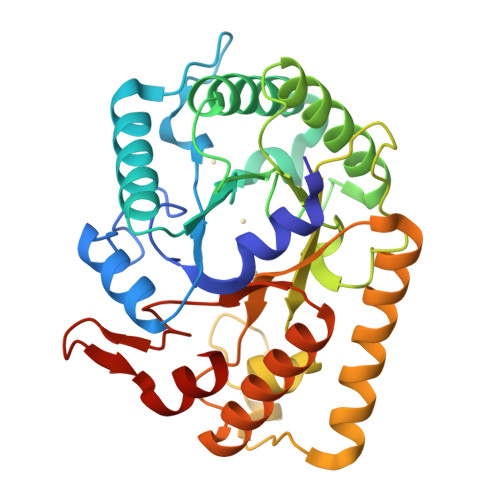
Crystal structure of endoglucanase Cel5A from the hyperthermophilic Thermotoga maritima
Tm_Cel5A, which belongs to family 5 of the glycoside hydrolases, is an extremely stable enzyme among the endo-acting glycosidases present in the hyperthermophilic organism Thermotoga maritima. Members of GH5 family shows a common (β/α)8 TIM-barrel fold in which the catalytic acid/base and nucleophile are located on strands β-4 and β-7 of the barrel fold. Thermally resistant cellulases are desirable for lignocellulosic biofuels production and the Tm_Cel5A is an excellent candidate for use in the degradation of polysaccharides present on biomass. This paper describes two Tm_Cel5A structures (crystal forms I and II) solved at 2.20 and 1.85 Å resolution, respectively. Our analyses of the Tm_Cel5A structure and comparison to a mesophilic GH5 provides a basis for the thermostability associated with Tm_Cel5A. Furthermore, both crystal forms of Tm_Cel5A possess a cadmium (Cd2+) ion bound between the two catalytic residues. Activity assays of Tm_Cel5A confirmed a strong inhibition effect in the presence of Cd2+ metal ions demonstrating competition with the natural substrate for the active site. Based on the structural information we have obtained for Tm_Cel5A, protein bioengineering can be used to potentially increase the thermostability of mesophilic cellulase enzymes.
Pereira JH, Chen Z, McAndrew RP, Sapra R, Chhabra SR, Sale KL, Simmons BA, Adams PD: Biochemical characterization and crystal structure of endoglucanase Cel5A from the hyperthermophilic Thermotoga maritima. J. Struct. Biol. 2010, 172:371-379PDB IDs: 3MMU [Diffraction data] 3MMW [Diffraction data]Raw diffraction data: 3MMU, 3MMW
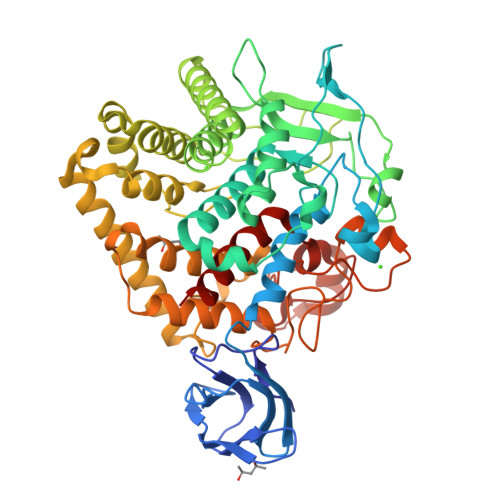
Crystal Structure of endoglucanase Cel9A from the thermoacidophilic Alicyclobacillus acidocaldarius
The production of biofuels using biomass is an alternative route to support the growing global demand for energy and to also reduce the environmental problems caused by the burning of fossil fuels. Cellulases are likely to play an important role in the degradation of biomass and the production of sugars for subsequent fermentation to fuel. Here, the crystal structure of an endoglucanase, Cel9A, from Alicyclobacillus acidocaldarius (Aa_Cel9A) is reported which displays a modular architecture composed of an N-terminal Ig-like domain connected to the catalytic domain. This paper describes the overall structure and the detailed contacts between the two modules. Analysis suggests that the interaction involving the residues Gln13 (from the Ig-like module) and Phe439 (from the catalytic module) is important in maintaining the correct conformation of the catalytic module required for protein activity. Moreover, the Aa_Cel9A structure shows three metal-binding sites that are associated with the thermostability and/or substrate affinity of the enzyme.
Pereira JH, Sapra R, Volponi JV, Kozina CL, Simmons B, Adams PD: Structure of endoglucanase Cel9A from the thermoacidophilic Alicyclobacillus acidocaldarius. Acta Cryst. 2009, D65:744-750.PDB IDs: 3EZ8 [Diffraction data]Raw diffraction data: 3EZ8