Automated Recommendation Tool (ART): A tool that leverages machine learning and probabilistic modeling techniques to guide synthetic biology in a systematic fashion, without the need for a full mechanistic understanding of the biological system.
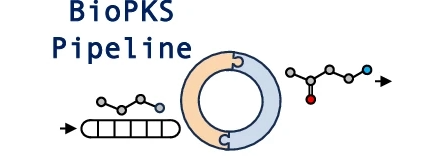
BiOPKS pipeline uses both multifunctional type I polyketide synthases (PKSs) and monofunctional enzymes for chemical biosynthesis. The platform is built upon two key rule-based components – RetroTide and DORAnet. When a user inputs a target structure, RetroTide first suggests chimeric PKS designs to synthesize the carbon backbone of this target. DORAnet then performs enzymatic post-PKS decorations of the PKS product to reach the target chemical. If an integrated PKS and post-PKS pathway design is found, the user can collect this design from BioPKS pipeline for future experimental synthesis in the laboratory.

ClusterCAD: A database and web-based toolkit designed to enable researchers to harness the potential of type I modular polyketide synthases for combinatorial biosynthesis. It provide tools to identify a truncated PKS as a starting point for domain exchange experiments given a small molecule target, and to select subunits as catalytic domain donors to alter a module’s acyltransferase, ketoreductase, or reductive cassette.

ICE: A cloud-based freely open-source repository with physical sample tracking capabilities that stores microbial strain, sequence, and plant seed information, including that associated with JBEI publications. Includes features such as DNA sequence editing and annotation, sequence validation results analysis, FASTA/GenBank/SBOL 2 support, and Web-of-Registries functionality.
VectorEditor: A cloud-based, freely open-source visual DNA editing and annotation tool.

DIVA: DIVA (Design, Implementation, and Validation Automation) improves the operational efficiency of companies and research institutions by decoupling DNA design from fabrication. The web-based software platform enables users to collaboratively and visually design DNA constructs and submit the designs to internal resources or outside vendors for construction at scale. Users can then track the construction and subsequent sequence validation processes using DIVA.
Experiment Data Depot (EDD): A cloud-based freely open-source experiment data repository that stores proteomics, metabolomics, transcriptomics, flow cytometry, plate reader, and BioLector digital data, including that associated with JBEI publications.

Technoeconomic Model Wiki JBEI is developing technoeconomic models of various aspects of lignocellulosic biofuel production. The models can be used to estimate the impact of various market and technology developments on the economics of biofuel and enzyme production. The models and the assumptions used to construct them have been deposited on the wiki, which allows users to choose the parameter scenarios that are of most interest to them. Read more.

RiceNet is a probabilistic functional gene network for 41,203 non-TE related genes of Oryza sativa (by TIGR release 5), constructed using a modified Bayesian integration of many different data types from several different organisms, with each data type weighted according to how well it links genes that are known to function together in Oryza sativa.

Rice Kinase Database The Rice Kinase Database was created to host functional genomic information gathered as part of an NSF funded rice kinase proteomics project and subsequently updated, expanded and maintained by JBEI.

Rice Oligonucleotide Array Database (ROAD) is a public resource for gene expression and coexpression analysis in rice, the most important staple food over the world. Over 1,000 microarray slides were incorporated and several kinds of analysis tools are available in ROAD.

JBEI GT Collection The JBEI GT Collection, the first glycosyltransferase clone collection specifically targeted for the study of plant cell wall biosynthesis, is expected to drive basic scientific understanding of GTs and better enable the manipulation of plant cell walls for the production of biofuels and other chemical products. The Arabidopsis clones are available from ABRC (abrc.osu.edu).

KitBase: KitBase contains detailed analysis of about 3000 mutant lines with mutations in ~40,000 genes. The resource was generated by fast-neutron-induced mutation in the model rice cultivar KitaakeX, and whole genome sequencing in collaboration with the Joint Genome Institute and Hudson Alpha.

MaxBin: a software package used for binning assembled metagenomic sequences, based on an Expectation-Maximization algorithm. MaxBin allows users to understand the underlying genomes of the microbes in their metagenomes by providing assembled metagenomic sequences and the reads coverage information or sequencing reads. MaxBin will report genome-related statistics, including the estimated completeness, GC content, and genome size. Users can then use MEGAN or similar software to find the taxonomy of each bin.
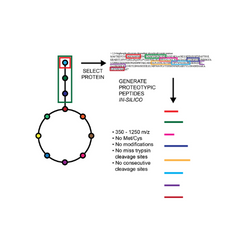
Targeted Proteomic toolkit for Escherichia coli proteins To expand the utility of quantitative proteomics for metabolic engineering, we validated and optimized targeted proteomics methods for over 400 proteins from more than 20 major pathways in E. coli metabolism (Batth TS, et al., 2014). Complementing these methods, we constructed a series of synthetic genes to produce concatenated peptides (QconCAT) for absolute quantification of the proteins and made them available through the Addgene plasmid repository.
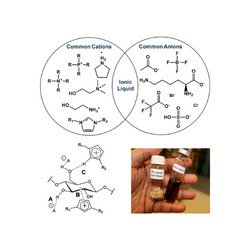
JBEI Ionic Liquid Database is a specialized research resource that consolidates critical data on ionic liquids, which are unique salt-based solvents used to process plant biomass for renewable fuels and chemicals. The database gives scientists extensive physicochemical attributes and performance data for various ionic liquids, which is critical for establishing the most effective and sustainable techniques for dissolving biomass. By facilitating the selection and development of these improved solvents, the tool considerably decreases the time and expense required to optimize the critical pretreatment stage in biorefining, enabling the scale-up of a more efficient and sustainable bioenergy industry.
JBEI Biositing Webtool is a public-facing geospatial platform that promotes long-term economic development and clean energy in the United States. This innovative resource combines data from a variety of sources, including agricultural resources, urban waste streams, and existing infrastructure, to assist policymakers, entrepreneurs, and researchers in making informed decisions.
JBEI Database of Lignin Modifying Enzymes is a research tool for accelerating the development of sustainable, renewable biofuels and bioproducts. This resource speeds up scientific discovery by cataloging and defining the important enzymes required to successfully break down lignin, the strong, protective substance found in non-food plant biomass. It enables researchers and business partners worldwide to overcome a fundamental technological barrier in bioenergy production, resulting in more efficient processes, lower prices, and a greater ability to use sustainable agriculture and forest waste to create a cleaner, more secure energy future.